
Spinal Muscular Atrophy Causes
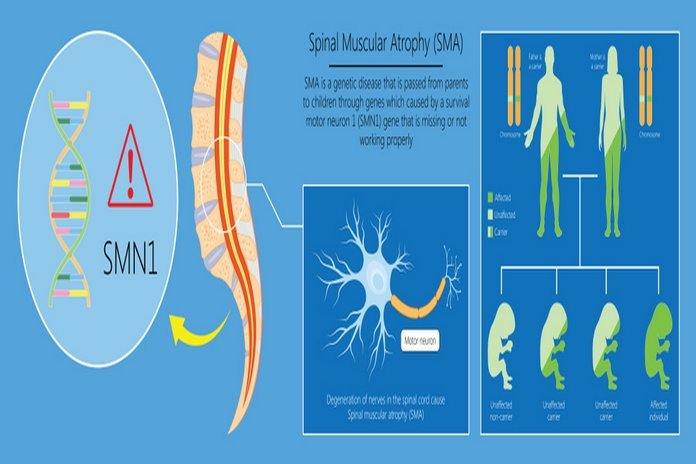
The error in both copies of the survival motor neuron one gene (SMN1) present on the 5q chromosome is the most apparent and prevalent form of spinal muscular atrophy. The SMN 1 gene is responsible for the production of survival motor neuron SMN protein, which maintains the standard functions and health of motor neurons. Some people with insufficient SMN protein levels develop spinal muscular atrophy. Insufficient SMN protein causes the loss of motor neurons in the spinal cord, resulting in atrophy and weakness of skeletal muscles. The weakness caused by spinal muscular atrophy is more pronounced in the upper leg and arm muscles, as well as the trunk, than in the muscles of the feet and hands.
Multiple alterations or mutations in the same genes result in multiple forms of spinal muscular atrophy. Some uncommon forms of spinal muscular atrophy result from mutations in other genes, including those on chromosome 20, chromosome 14, and chromosome 9. The variety of spinal muscular atrophy is determined primarily by the severity of muscle weakness and the age of onset. Nonetheless, there is perpetual overlap between the various varieties of SMN.
SMN and Genes
Spinal muscular atrophy is typically inherited in an autosomal recessive manner, meaning that the affected individual possesses two mutated genes. It is typical for each patient with SMN to inherit each mutated gene from both parents. However, in a few rare instances, SMN is caused by mutations in the UBA1 gene. People who possess a single mutated gene do not exhibit any symptoms of the disorder, but they are disease carriers. As SMN is an autosomal recessive disorder, it is possible that it affects more than one member of the same family.
Under normal circumstances, the SMN1 genes generate entirely functional and full-length SMN proteins, which are essential for the body’s neurological and muscular functions. When the SMN1 alleles are mutated, insufficient amounts of SMN proteins are produced. As a result of the chromosome 5 mutation, the adjacent SMN2 gene is also affected. The proteins produced by the SMN2 genes are produced in minute quantities and are completely inactive. Moreover, only 10 to 15% of the functionality of SMN proteins remains.
Multiple copies of the SMN2 gene are located within the human body. Typically, each individual has zero to eight copies of SMN2 genes. The greater the number of copies of SMN2 genes in the body, the more functional SMN protein is available in the body; as a result, the severity of the disease symptoms is greatly reduced. The severity of the disease also depends on the onset and severity of various biological pathways that exert an influence. The biological modifiers, such as the ZPR1 and plastin-3 proteins, indicate the onset or severity of the disease.
.){kind=link}